Trisomia 13. Presentación de un caso.
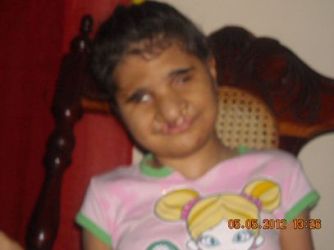
Paciente de 13 años, mestiza, residente en comunidad urbana “Micro 70”, sin problemas de tipo social y de higiene ambiental, con antecedentes patológicos personales de bajo peso al nacer. Estando la madre embarazada a término y con un embarazo aparentemente normal se detecta por ultrasonido, malformación renal y se remite a Ciudad Habana, Hospital Maternidad Obrera donde se produce el parto por cesárea. En el Servicio de Neonatología de dicho hospital, se detectan malformaciones al examen físico como labio leporino y paladar hendido, hipoplasia de globos oculares, estreches de las hendiduras palpebrales, implantación baja de las orejas, ausencia de cuero cabelludo, cejas y pestañas, dedos de las manos en flexión y ausencia parcial de huesos parietales. Después de un adecuado examen físico, el pediatra hace el diagnóstico presuntivo de un recién nacido pretérmino, bajo peso con malformaciones congénitas. La paciente es ingresada por 21 días en Ciudad Habana, se traslada a sala de prematuros del Hospital Héroes del Baire en la Isla de la juventud y después de una tórpida y complicada evolución es dada de alta con 3 meses de de nacida. Lo más curioso de este caso es que la esperanza de vida de estos pacientes es casi nula y sin embargo esta paciente ha sobrevivido todos estos años.
Patient the 13 years, it crossbreeds, residing in urban community “Micro 70″, without problems of social type and of environmental hygiene, with APP of under weight when being born. Being the pregnant mother to term and with a seemingly normal pregnancy it is detected by ultrasonido, renal malformation and it is remitted to City Havana, Hospital Labor Maternity where the childbirth takes place for Caesarean operation. In the Service of Neonatología of this hospital, malformations are detected to the physical exam as harelip and cracked palate, hipoplasia of ocular globes, narrow of the fissures palpebrales, low installation of the ears, absence of hairy leather, you slack and lashes, fingers of the hands in flexion and partial absence of parietal bones. After an appropriate physical exam, the pediatrician makes the presumptive diagnosis of a newly born pretérmino, under weight with congenital malformations. The patient is entered by 21 days in City Havana, she moves to room of premature of the Hospital Heroes of the Baire in the youth’s Island and after a torpid and complicated evolution she is given of high with 3 months of born. The most curious in this case is that the hope of these patients’ life is almost null and however this patient has survived every year.
Las alteraciones más frecuentes del número de cromosomas son las trisomías. Aparecen cuando existen tres representantes de un determinado cromosoma en lugar de los dos habituales. Estas suelen ser el resultado de una no disyunción meiotica (falta de separación de un par de cromosomas). 1
La prevalencia de la trisomía 13 es de aproximadamente 1:12.000 nacidos vivos. Cuanto mayor sea la pareja, más probabilidad tienen de engendrar un hijo que presente dicho síndrome. El riesgo de recurrencia (de tener un segundo hijo con síndrome de Patau) es bastante bajo en el caso de que ningún padre presente la translocación.2 3
El síndrome de Patau, también conocido como trisomía en el par 13, Trisomia D o síndrome de Bartholin-Patau, es una enfermedad genética que resulta de la presencia de un cromosoma 13 suplementario (consecuencia de una no disyunción meiotica, principalmente en el gameto materno). Aproximadamente un 20% de casos se deben a traslocaciones, siendo la t (13q14) la más frecuente. Sólo un 5% de dichas traslocaciones es heredada de uno de los progenitores. En el caso de la translocación, aunque los padres estén sanos tienen posibilidad de pasar la enfermedad a su descendencia. Los mosaicos representan 5% de los casos de trisomía 13. 4
Este síndrome, es la Trisomia reportada menos frecuente en la especie humana. Fue observado por primera vez por Thomas Bartholin en 1657, pero no fue hasta 1960 cuando la descubrió Patau. Los afectados por dicho síndrome pueden morir poco tiempo después de nacer, la mayoría a los 3 meses, y como mucho llegan al año. Se cree que entre el 80-90% de los fetos con el síndrome no llegan a término. Los fetos afectados de Trisomia 13 presentan anomalías múltiples que pueden ser detectadas antenatalmente por medio de la ecografía, el diagnóstico se confirma a través de estudio cromosómico. 5 6
La prevalencia de la trisomía 13 es de aproximadamente 1:12.000 nacidos vivos. Cuanto más mayor sea, más probabilidad tiene de engendrar un hijo que presente dicho síndrome. El riesgo de recurrencia (de tener un segundo hijo con síndrome de Patau) es bastante bajo en el caso de que ningún padre presente la translocación. 6
El tratamiento de los síntomas casi siempre es personalizado. Se basa, sobre todo, en el tratamiento de las anomalías físicas que presenta el niño al nacer. Aún así, los recién nacidos con la trisomía 13 suelen precisar de asistencia médica desde el mismo momento de su nacimiento, ya que en 2 de cada 3 casos obtienen puntuaciones inferiores a 7 en el test de Apgar al primer minuto, descendiendo a los cinco minutos de vida. Debido a que las anomalías cardiacas representan la causa principal de mortalidad en los pacientes con síndrome de Patau (no suelen pasar las semanas de vida), existe un dilema ético sobre si la reparación quirúrgica de dicho sistema está indicada, teniendo en cuenta el pésimo pronóstico del cuadro desde el punto de vista físico e intelectual. Los padres deben, por su parte, conocer determinados cuidados que tendrán que llevar a cabo en los hijos con el síndrome, ya que pueden ser de importancia vital para la supervivencia de los mismos. 7 8
Paciente de 13 años, mestiza, residente en comunidad urbana “Micro 70”. Nacida producto de un parto distócico el 19 de junio de 1998 por cesárea a las 39 semanas con llanto fuerte y apgar 9-9 al nacer con un peso de 2290 gramos, talla 47 cms y CC de 28,5 cm y caída del cordón umbilical a los 30 días.
En el Servicio de Neonatología de Maternidad Obrera, se detectan malformaciones al examen físico como labio leporino y paladar hendido, hipoplasia de globos oculares, estreches de las hendiduras palpebrales, implantación baja de las orejas, ausencia de cuero cabelludo, cejas y pestañas, dedos de las manos en flexión y ausencia parcial de huesos parietales. Después de un adecuado examen físico, el pediatra hace el diagnóstico presuntivo de un recién nacido pretérmino, bajo peso con malformaciones congénitas.
Ingresa al Servicio de Neonatología procedente de Ciudad habana a los 21 días de nacida con el diagnostico de bajo peso al nacer y trisomia 13 o síndrome de Patau donde se mantiene hasta recuperar el peso.
Ha requerido ingreso hospitalario por:
Complicaciones neurológicas.
EDA.
Piodermitis.
Neumonías a repetición.
Absceso en muslo por vacunación.
Intolerancia a los lácteos.
Desnutrición proteica energética.
Enfermedad respiratoria a repetición.
Síndrome febril.
Estatus Convulsivo.(epilepsia)
Antecedentes patológicos familiares:
Madre viva y sana
Padre con familiares malformados.
Examen físico general:
Paciente longilinea que no deambula y guarda decúbito activo indiferente con fascies inexpresiva.
Mucosas, piel, tejido celular subcutáneo: normal.
Peso corporal: 29kg
Talla: 139 cm
Pelos y uñas: sin alteración.
Examen físico regional
Cabeza alargada y flexionada hacia delante la mayor parte del tiempo.
En la cara se observa labio leporino y paladar hendido, hipoplasia de globos oculares, estrechez de las hendiduras palpebrales, implantación baja de las orejas.
Tórax: Propio de su biotipo sin retracciones y abombamientos.
Abdomen: Plano con hernia umbilical marcada y que sigue los movimientos respiratorios y los golpes de tos.
Extremidades:
Dedos de las manos y los pies en flexión.
Examen físico por aparatos.
Aparato respiratorio: Normal.
Aparato Cardiovascular: Ruidos cardiacos rítmicos, con soplo sistólico de eyección grado III/ VI en mesocardio.
Sistema digestivo: No control de esfínter anal.
Alimentación asistida, come de todo.
Sistema genito urinario: No control de esfínter vesical.
Sistema Hemolinfopoyetico: Sin alteraciones.
Sistema nervioso: Paciente consciente, con ausencia total del lenguaje con fascies inexpresiva.
Trofismo Normal.
Reflectividad: conservada.
Tono muscular conservado.
Motilidad
Movimientos voluntarios: presentes.
Movimientos involuntarios: cuando convulsiona.
Coordinación estática y dinámica alterada.
Praxia: Alteraciones de la palabra hablada (afasia), de la palabra escrita (agrafia) de la mímica (apraxia).
Sensibilidad superficial Táctil, térmica y dolorosa sin alteraciones.
Sensibilidad profunda: Ausente.
Tratamiento médico actual.
1. Dieta por síndrome de malabsorcion.
2. Clonazepan (1mg) 1 tab cada 12 horas.
3. Convulsin (50 mg) 1 tab cada 6 horas.
Este caso, con pronóstico muy desfavorable de vida, que después de su nacimiento y evolución posterior se le puso el nombre de “Milagro” ha sido un ejemplo más del desarrollo de la medicina cubana y del amor y cariño con que ha sido cuidada en estos trece años. Es importante recalcar el cuidado esmerado de su familia en especial su madre que ha mantenido una actitud positiva y abnegada, así como el equipo básico de salud del consultorio y el equipo multidisciplinario de especialistas del Hospital Héroes del Baire que también han contribuido muchísimo ha mejorar la calidad de vida de esta paciente.
1 Tratado de pediatría 15.a edición Volumen 1 Capitulo 67. 394-397.
2 Dave McDonald. Cytogenetics Information Site (ed.): Citogenética básica. ( 2011).
3 Feuk L., Carson A. R. y Scherer S. W. «Structural Variation in the human Genome.». Nature Reviews Genetics 7 (2): pp. 85-97(2006).
4 Gilbert F, Chromosome 13. Genet Test 4 (1): pp. 85-94. (2000).
5 Kivela T, Tuppurainen K, Riikonen P, Vapalahti M (Retinoblastoma associated with chromosomal 13q14 deletion mosaicism. Ophthalmology 110 (10): pp. 1983-8. 2003).
6 Dunham A, Matthews LH, Burton J, Ashurst JL, Howe KL,et al. The DNA sequence and analysis of human chromosome 13. Nature 428 (6982): pp. 522-8. (2004).
7 Novo Villaverde, F.J. Genética Humana. Madrid: Pearson. (2007).
8 The ENCODE Project Consortium. Identification and analysis of Functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 (7146): pp. 799-816. (2007).
Anexo 1.